Objectives
At the end of the session students should know what NMR can be used for and how to prepare a sample for an initial NMR spectrum
Texts
A good textbook for macromolecular NMR with a mixture of theory and results
Jeremy N.S. Evans Biomolecular NMR Spectroscopy Oxford University Press 1995 ISBN 0 19 854766 8
A simpler practical introduction to macromolecular NMR is found in
NMR of macromolecules: A practical approach ed GCK Roberts Oxford 1993
A much more mathematical treatment of theory is given in
Protein NMR Spectroscopy J. Cavanagh, WJ Fairbrother, AG Palmer III, NJ Shelton. Academic Press 1996
![]() Introduction to
NMR Spectroscopy
Introduction to
NMR Spectroscopy
![]() Structure
determination by NMR
Structure
determination by NMR
![]() Why you
should get an NMR spectra on most proteins
Why you
should get an NMR spectra on most proteins
![]()
Introduction to NMR Spectroscopy
Nuclear magnetic resonance depends on the quantum mechanical property of nuclear spin. This is quantised spin angular momentum, which confers a magnetic moment on a nucleus in a magnetic field. The nuclear spin (I) can have a value of 0, 1/2, 1, 3/2, 2 etc. Each nuclear spin I has 2I+1 states given by the quantum number m -I, -I+1 ....I-1, I. So for I=1/2 nuclei like 1H the states are m=-1/2 and m=+1/2. This kind of nucleus can be thought of as a bar magnet, which can be aligned with or against the magnetic field. At this point the text books resort to classical mechanics to derive the following equation
ω=γBo
γ is an intrinsic property of the nucleus (the quadropole moment), ω is the Larmor frequency and is the absorption frequency of an isolated atom. NMR spectrometers are normally described in terms of the Larmor frequency of 1H in their magnetic field. These are say 200 or 300 MHz for organic chemists routine spectra, 500, 600 Hz and increasingly 750 and 800 MHz are the work horses of macromolecular work and 900 MHz are the virility symbols of the big macromolecular research groups at present and 1GHz machines are being planned. 800 MHz corresponds to 18.8 Tesla, which is an extremely expensive magnet (several hundred thousand pounds), but the NMR people would argue is peanuts compared to a synchrotron beam lines used by the X-ray crystallographers. There is a Boltzmann energy distribution between the parallel and anti-parallel states given by
Nb/Na=e(-γ ħBo/kT)
Energy is put into the system by radio waves from the spectrometer, which displaces the distribution of the system away from equilibrium and then the decay back to equilibrium is monitored by the emitted radio waves.
![]()
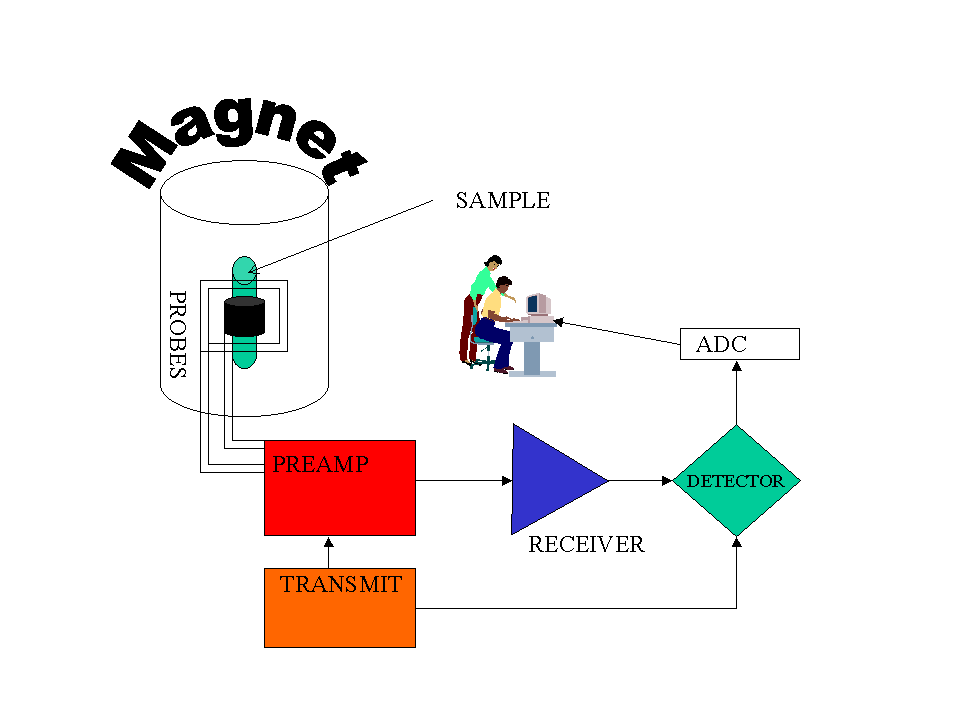

Figure 1 (above) shows a schematic of a NMR spectrometer. The main components are the magnet,
which is normally a liquid nitrogen cooled electromagnet, the probes (three or even four probes
which are coils surrounding the sample are common), the electronics to transmit and receive the
radio waves and finally a computer via an analogue to digital convertor.
Figure 2 (left) shows a picture of the National 800MHz spectrometer in Cambridge (Taken from http://www.bio.cam.ac.uk/nmr/800/index.html)
![]()
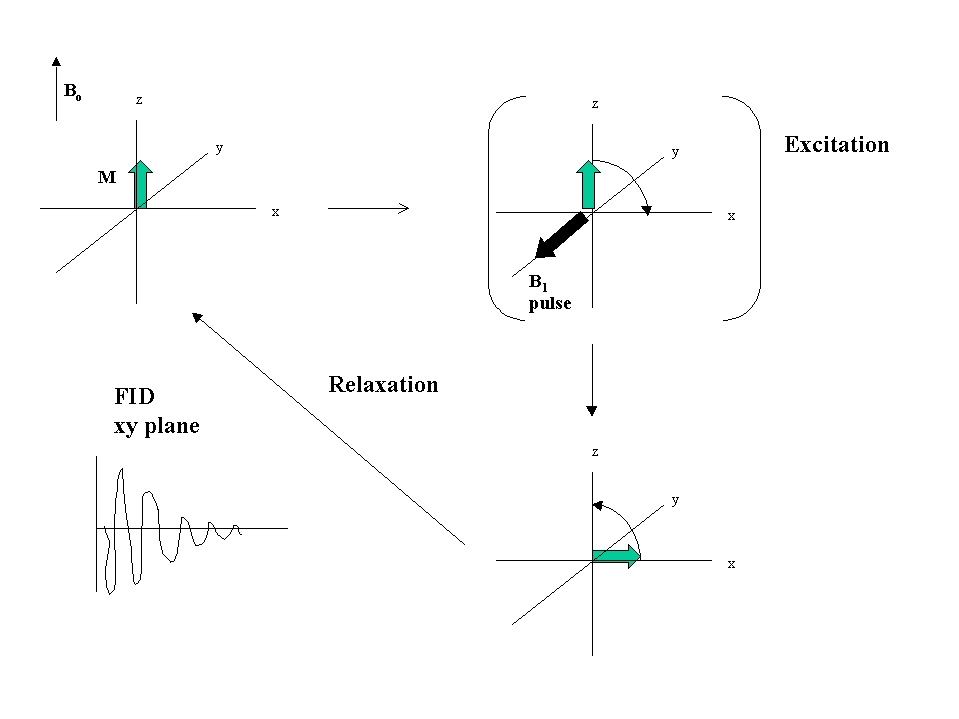
A single short pulse is applied containing frequencies over the spectral width required. This is of the order of 10-5 s. The nuclear spin then decays back to equilibrium and the decay is followed. Once it has decayed the process can be repeated and summed to improve the signal to noise. The decay is digitised and a Fourier transform applied to convert the so called Free Induction Decay (FID) to a frequency spectrum. The conventional representation of this is by a vector representation (see Figure 3 below).
Having summed several pulses and carried out a Fourier Transform you obtain a 1D spectrum. Each proton has a chemical shift (Measured in parts per million ppm) which is given by
δ=(ω-ωTMS)/ωox106
Where ωo is the operating frequency of the spectrometer and ωTMS is the proton frequency from tetramethylsilane. The value of the chemical shift depends on the local magnetic field experienced by that proton, which is related to its chemical environment. Obviously if there are protons in the solvent the biggest peak comes from the solvent. Amide NH protons exchange with the solvent and so if a protein is left in D2O there will be no signal from the amide protons. Some spectra to look at the side chain atoms are best done in D2O but the NH proton is very important in determining the backbone of the protein and so most experiments are carried out in 90% H2O/10% D2O. The 10% D2O is required as a lock signal is obtained from the D2O which is monitored to keep the magnetic field homogeneous. Extra pulses are included to reduce the size of the water peak. (NMR spectroscopy is full of acronyms for pulse sequences. Solvent suppression sequences include WATERGATE and DANTE.) The water peak divides the spectrum into two regions above the water peak (4.7 ppm) are the aromatic and NH protons below the peak are the aliphatic protons. OH protons are usually too broad to be seen. A typical protein spectra is seen in Figure 4.
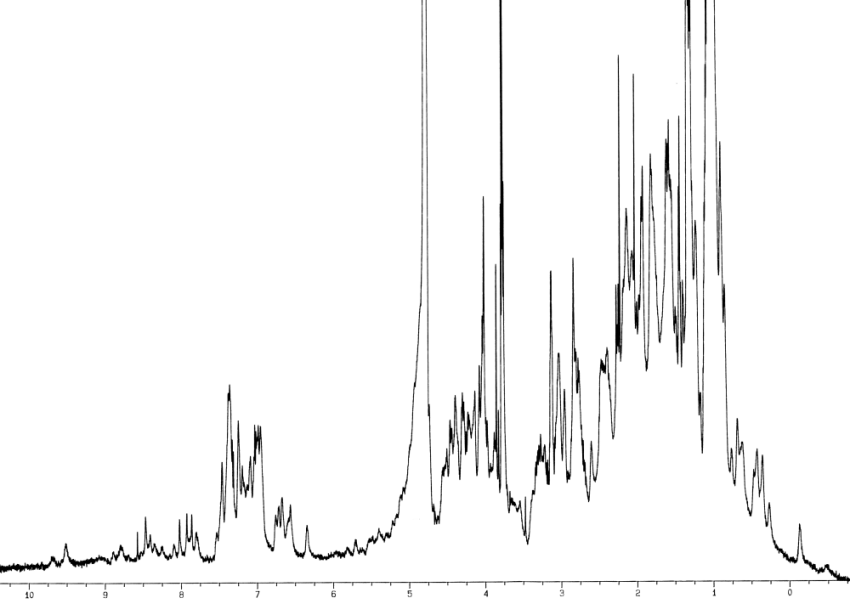
Figure 4 600MHz spectrum of N terminal domain of p47phox
![]()
The major natural isotopes of carbon and oxygen 12C and 16O have I=0 and so are NMR inactive, 14N has I=1, but this relaxes much faster than spin 1/2 nuclei and so does not have an observed effect in macromolecular NMR. However 13C and 15N have I=1/2. These can be incorporated at nearly 100% abundance by growing cells on isotope enriched media. For 15N this is not too bad (£50 a litre for E.coli minimal media), for 13C it is £1000 a litre (although prices are edging down as use increases). This means you need very good expression (the yield normally drops on going from a rich media like 2TY or TB to minimal media). There are isotopically enriched media available for Pichia and methyltrophic yeasts. In theory you should be able to record 13C and 15N spectra directly. In fact better spectra are obtained by detecting the signals indirectly via the much more sensitive 1H spins. This is done by the so called INEPT pulse sequence. 2H which is a quadropolar (I=1) nuclei can also be incorporated. Partial deuteration has advantages (which I will not attempt to explain) in pushing the size limits of NMR. As the isotope effect of deuterium has a significant effect on the metabolism of the bacteria, slow conditioning in increasing D2O concentrations are required. Incorporation of specifically labelled individual amino acids eg 15N His or 13C Ala can be done. There is not a need to use auxotrophs if it is done in the presence of the other amino acids in the anabolic pathway. These can produce simpler spectra for helping interpret fully labelled spectra or for probing particular amino acids (eg the protonation state of a His in an enzyme).
![]()
If all you could obtain were the chemical shifts of every C, H and N atom in the protein, there is a formal possibility that you could determine the structure by calculating the chemical shift of each nuclei from a model and altering the model to improve the fit. This is not yet possible, although some people are trying to use chemical shift calculations as part of a structure determination. However there is interaction between spins, which can be used and detected. There are basically two types of spin coupling; through space (dipolar) and through bond. (scalar, correlation or J coupling). Through space coupling which is also known as the Nuclear Overhauser Effect (NOE) is dependent on r-6, which means that signals can be got between protons that are less than 5Å apart. The simplest experiment produces a cross peak at the chemical shifts of pairs of protons that lie less than 5 Å apart. This is the basis of NMR structure determination, which consists of calculating a model compatible with most (if not all) the observed NOE's in the spectrum. However before this can be done each NOE has to be assigned to a pair of protons in the protein, so the chemical shift of each proton needs to be known. This is done by assembling 'spin systems' which are protons which are linked through bonds. These correspond to amino acids. Connections then need to be made to adjacent amino acids.
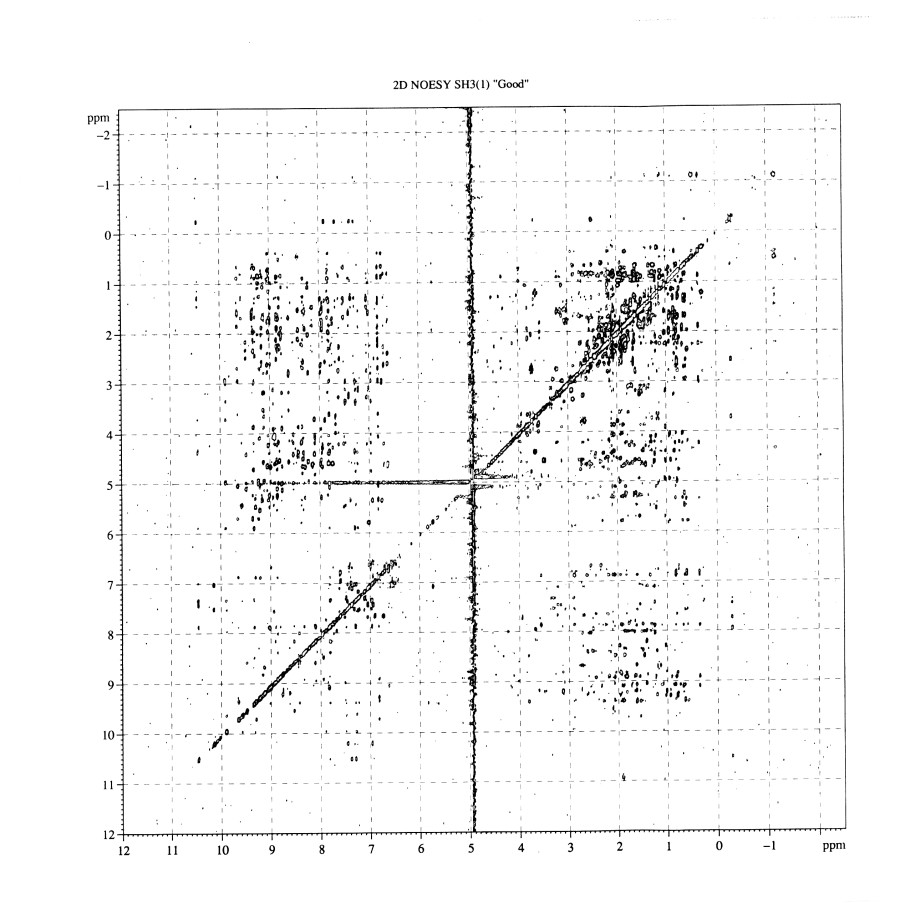
Figure 5 2D NOESY spectra. Click here for close up of a region
{kind=link}
![]()
For proteins of less than 15kD the structure can be determined by proton experiments alone. One strategy is first to determine the spin systems in D2O using 2D through bond experiments (DQF-COSY and TOCSY). Then from experiments in H2O, the exchangeable protons are added to the spin systems. Sequential spin systems are identified by looking for NOE's between the NH proton and the previous residue (so called dNN, dαN,dβN) links see Figure 6. The pattern of these links are indicative of certain types of secondary structure and the secondary structure is obtained sometime before the structure is fully solved. Secondary structure assignments are publishable and are the NMR equivalent of crystallisation notes, indicating that the structure is solvable.
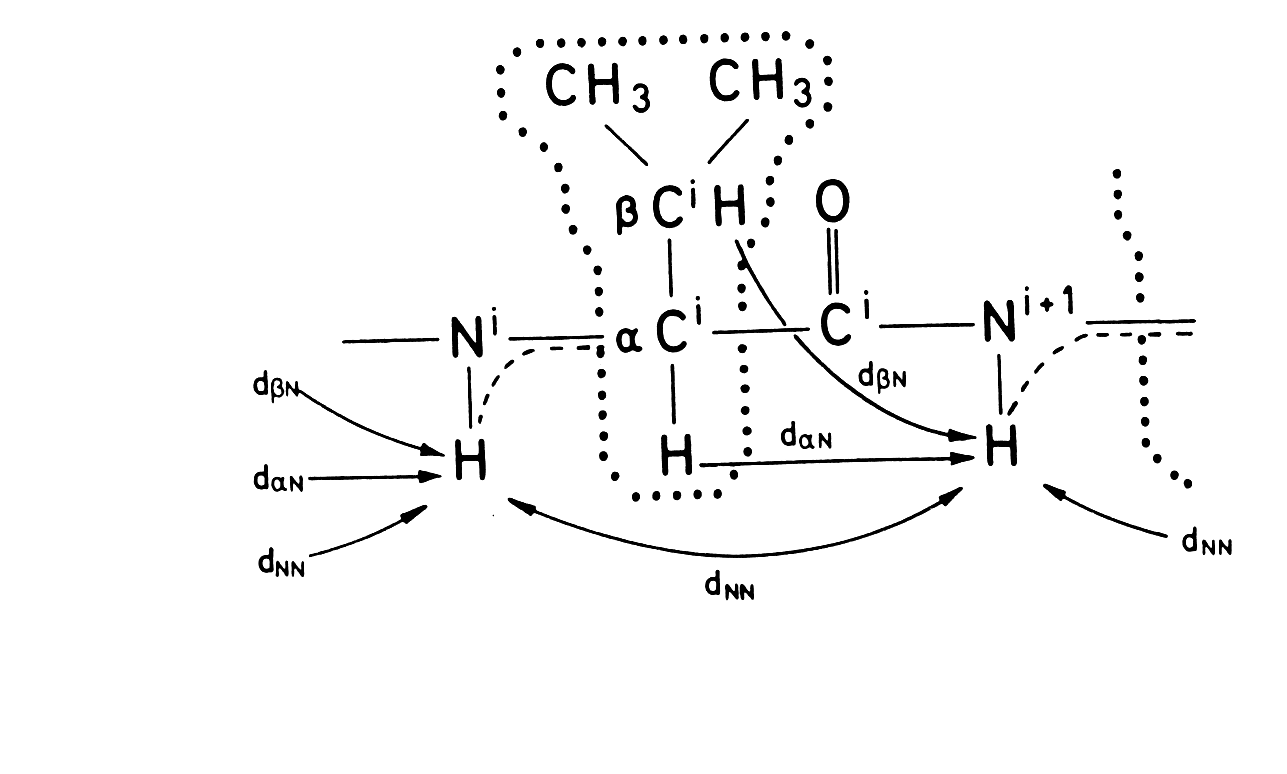
Figure 6. HH distances in an amino acid. Taken from Evans Fig 4.1
For proteins of 15-30kD the 2D spectra are too crowded to interpret and the proton chemical shifts alone are ambiguous as to which proton in the protein they are. For proteins in this range isotopically labelled samples are required and 3 or 4D experiments are carried out which attach a 13C or 15N shift to each proton as well. A 3D experiment is a stack of 2D experiments but each peak only occurs once in the stack so there are many less peaks per 2D layer. Assignments are carried out by a series of experiments such as HNCO, HNCA, HN(CO)CA, HCACO, HBHA(CO)NH. There are 3 and 4D versions of the NOE and TOCSY experiments as well. There are complications to many of these experiments, which mean that not all the expected appears appear or extra peaks appear. For example in the HNCA experiment the CA of the previous amino acid is often also visible. 4D experiments in theory would resolve all the protons in large proteins. However there are constraints on the relationship of line width and tumbling time that means that even with a number of recent improvements such as partial deuteration the limit is likely to be reached under 40kD. Solid state methods and a new method for determining bond dipoles in a protein partially orientated in the magnetic field offer possibilities for pushing the size limit.
What is obtained from in an NMR structure determination is a list of distance constraints (ie that certain protons are less than a certain distance apart). There is some correlation of distance with intensity but normally the uncertainty is several Å. These are combined with the stereochemical constraints (bond distances, bond angles etc) of proteins similar to those used in X-ray crystallography. The best solution to these restraints are found using distance geometry or molecular dynamics software (CNS, X-PLOR, DISMAN, DIANA). It is normal to carry out these calculations tens of times, collect together those that pass some quality test on satisfying the constraints and display them together (Figure 6). This gives a much clearer idea of which parts of the structure are well defined than normally given by X-ray. Sometimes an average structure is calculated, but this will normally have poor geometry and in that sense is less right than the individual solutions. The database OLDERADO at the University of Leicester analyses these clusters and among other information tells you which of the models is the most typical. This is perhaps the best model to then look at in display programmes.
There are also NMR experiments that allow constraints on certain bond angles to be added and hydrogen bonds can be detected and used as restraints. A method has been described (Tjandra, N. and Bax, A (1997). Science 278:1111-1114) that obtains the angles between a bond (or more precisely the interatom vector) and the principal axes of the molecule. This requires that a molecule is not perfectly spherical (they rarely are) and that there is some ordering of the molecules in the magnetic field. This is achieved with a low concentration of a liquid crystalline lipid phase.
Significant increase in the range of proteins that can be studied has recently been achieved by
using deuteration of the protein. Deuterium is not NMR active. Normally the other hydrogens in a
large protein are a major source of the rapid decay. E.coli cells can be got to grow in
D2O and perdeuterated glucose or acetate. The exchangeable (NH) hydrogens are changed to
protons on transferring the sample to H2O and a structure can be determined based just on
the backbone hydrogens. More detail can be obtained by adding amino acids such as valine with
hydrogens on the methyl groups to the culture in D2O.
(For an example of a 280 amino acid protein done by Steve Matthews at Imperial see Nature
Structural Biology, 1999, Vol.6, No.4, Pp.313-318)
In 2002 the pulse sequences proposed in the last couple of years have started to produce biological results. These include transverse relaxation-optimised spectroscopy (TROSY), which reduces the speed of decay of the spin caused by adjacent atoms and cross correlation relaxed-enhanced polarisation transfer (CRINEPT) (Riek, Pervushin and Wuthrich (2000) TIBS 25:462-467). Using these spectra of GroES a 10kD protein has been collected from within the 72kD homoheptamer or even the 900kD GroES/EL complex. (Fiaux et al., Nature 418:207-211) The p53 core domain has been shown to be unfolded when bound to the 200kD Hsp90 complex (Rüdiger et al., (2002) PNAS 99:11085-11090. Thus these techniques allow spectra to be collected from large protein complexes, particularly if only small parts can be labelled to reduce peak overlap.
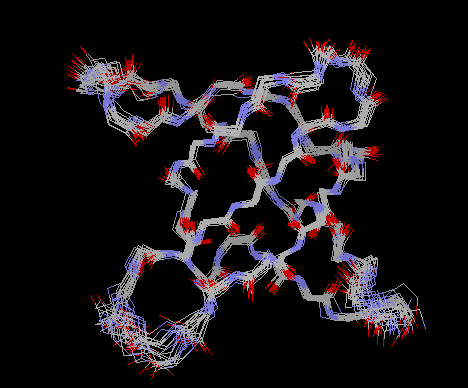
Figure 7 Backbone of well-defined set of NMR structures. The SH3 domain of fyn (PDB 1NYG). (CJ
Morton , DJ Pugh , EL Brown , JD Kahmann , DA Renzoni , ID Campbell "Solution structure and peptide
binding of the SH3 domain from human Fyn." Structure, 4 (6), 1996, 705-714)

Figure 8. Backbone of a less well superimposed set of structures PDB 1CYP(Kilby PM, Van Eldik LJ,
Roberts GC, Structure 1996 4(9):1041-52. The solution structure of the bovine S100B protein
dimer in the calcium-free state.) It is worth noting that in this case there are more restraints
(1773) than for the SH3 above (669). The greater spread of structures is due partly to the structure
being of an apo (calcium free) form of a calcium binding protein, which is inherently more flexible.
This structure is a dimer so there is a problem resolving those NOE's within and those between
chains.
![]()
Comparison of X-ray and NMR structure determination
|
Factor |
X-ray |
NMR |
Comment |
|
Sample in General |
Must form crystals |
Must be homogeneous, monodisperse, stable. |
The criteria for a NMR sample are thought to be the main criteria for growing crystals. NMR structures have been determined before the X-ray structures but there are few proteins that have not subsequently given crystal structures. |
|
Concentration |
Normally 2 mg/ml |
Must be greater than 0.2 mM ie about the same |
|
|
Temperature |
Anything above freezing |
As hot as possible. |
NMR spectra get much sharper with temperature even 30oC compared with 25oC can make a difference. |
|
Buffer |
Phosphate is bad for screening as it gives crystals with divalent cations |
Phosphate is the normal buffer as it is proton free and the right range |
|
|
pH |
Whatever works |
As low as possible to slow proton exchanges and strengthen H bonds. 5.0-6.0 is normal |
|
|
Molecular Weight |
1,000,000 |
30,000 |
Sometimes regions of a larger protein are unusually flexible and can be seen. Pyruvate dehydrogenase is the classical example. (Perham, Duckwork and Roberts, (1981). Nature 292, 474-477. |
|
Data collection |
There is only one type of final experimental restraint that is the F values from the highest resolution data set you have. Normally these days this is a single data set |
Several different experiments give the distance restraints and further experiments can be done to improve a structure with bond angle restraints, dipolar restraints etc. |
|
|
Structure Solution |
Several methods depending on problem (MIR, MAD, MR) |
There is no real equivalent of Molecular replacement ie it is not much easier to do a homologous structure |
You can assign point mutations by comparing NMR spectra and you can often assign most of a complex from the component assignments |
|
Final structure |
One set of coordinates with B factors |
An ensemble of coordinate sets |
Both methods have made mistakes and there have also been a few real differences. Neither is a physiological state. |
![]()
Flexibility
The decay of a signal (the relaxation) is linked to the overall tumbling of the molecule and the relative mobility of that particular atom. Crystallographic B factors do not distinguish between static and dynamic disorder as well.
Aggregation
The dependence of the spectrum on concentration gives information on aggregation (and allows a search for reagents that counteract it).
Folding
Protein folding and unfolding can be followed by changes in the NMR spectra with temperature or time. Stopped flow methods can be used to monitor exchange of protons. Unfolded protein is mixed with D2O. After a certain time the pH is dropped which prevents further exchange, but allows folding to continue and the more 1H left at a site the quicker that site is folded.
Ligand binding
Changes in spectra on addition of ligands can be used to show binding and to measure stoichiometry.
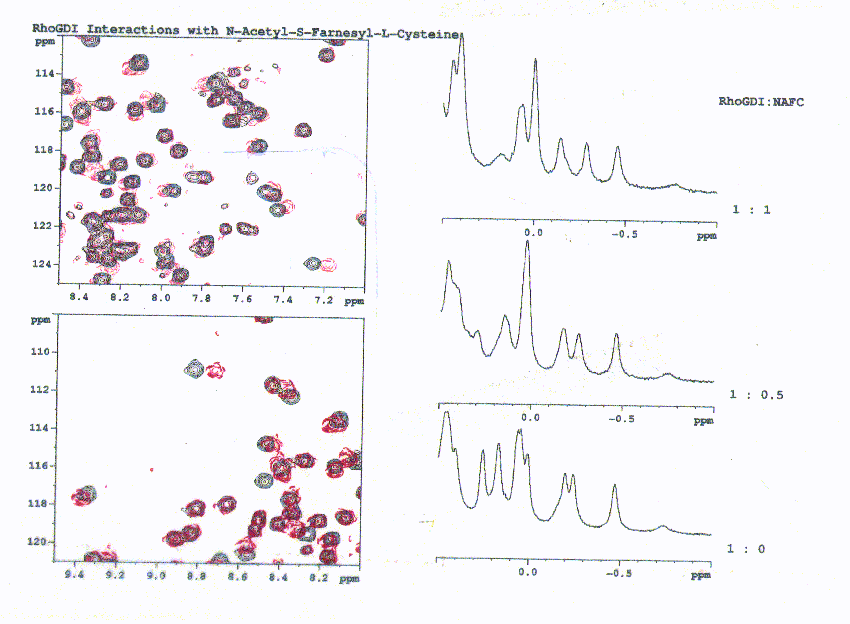
Figure 9. Changes in the 1D and 2D spectra on addition of N-acetyl-S-farneyl-L-cysteine to rhoGDI. The changes stop when a 1:1 ratio is reached indicating the stoichiometry
For ligands that exchange on and off a protein faster than the relaxation rate transferred NOEs (TRNOE) can be used to determine the bound structure. This is largely independent of the ligand size.
The binding of ligands can be detected at reasonably low affinity and so a method for drug design has been proposed by screening small molecules for low affinity binding by NMR and then assembling them into a more complex drug known as SAR (Structure Acticity Relationship)by NMR (Shuker et al., 1996 Science 2741531-1534).
Complexes
If one protein is isotopically labelled and the other is not, the residues that change chemical shift are implicated in binding. This is only really informative if you have assignments and ideally a structure. However this can be used to determine a binding constant and will detect specific interactions with very weak binding constants (up to mM)
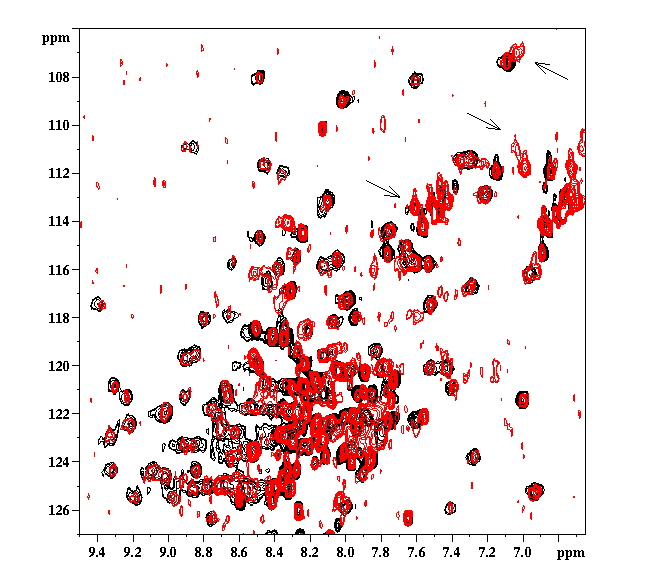
Figure 10. The 15N-1H HSQC spectra of intact RhoGDI (Black) and in the presence of equimolar rac1 (red). Examples of resonances affected by rac binding are indicated by arrows.
Enzyme Kinetics and Mechanism
NMR is a very good method for following enzyme kinetics and mechanism, particularly stereochemistry using stereospecific isotopic substrates. NMR is particularly good at determining the protonation state of residues.
In vivo
NMR is basically the same as MRI, which is increasingly used as an alternative to X-rays for body imaging. You can also follow metabolic fluxes in vivo and measure things like ATP concentration. This is a subject in itself.
Solid-state NMR
The sample must be spun at the 'magic angle' of 54o44' (3cos2θ -1=0). This is
normally combined with TOSS (Total sideband supression) to produce acceptable linewidths. The main
use has been in the studies of lipid bilayers and membrane proteins. The most notable success has
probably been to determine a gramicidin structure in a bilayer and it has given spetroscopic
information on a number of systems such as bacteriorhodopsin.
![]()
Why you get an NMR spectra on most small to medium sized proteins
If you have a protein under 50 kD (possibly even under 100 kD), it would be worth getting an initial spectra possibly even an 15N spectra. Lack of signal dispersion could be an indication of unfolded protein. Some sharp peaks would indicate an unfolded/unstructured region. This information cannot be got from CD. Variation of the spectrum with concentration would indicate aggregation. To prepare a sample, the protein should be purified in 20mM phosphate buffer pH between 4.0 and 6.0 (as low as possible) and concentrated to ideally around 1mM. 0.5 ml is normally the smallest sample volume. You must include 10% D2O. An example of NMR informing a crystallisation is work I did on rhoGDI. This is a protein of 204 amino acids. When an NMR sample was obtained there were two sets of peak line widths. This is shown both in 15N-1H HSQC spectrum and in 1D proton spectra. By removing the disordered region (by proteolysis) we obtained crystals of the folded region.
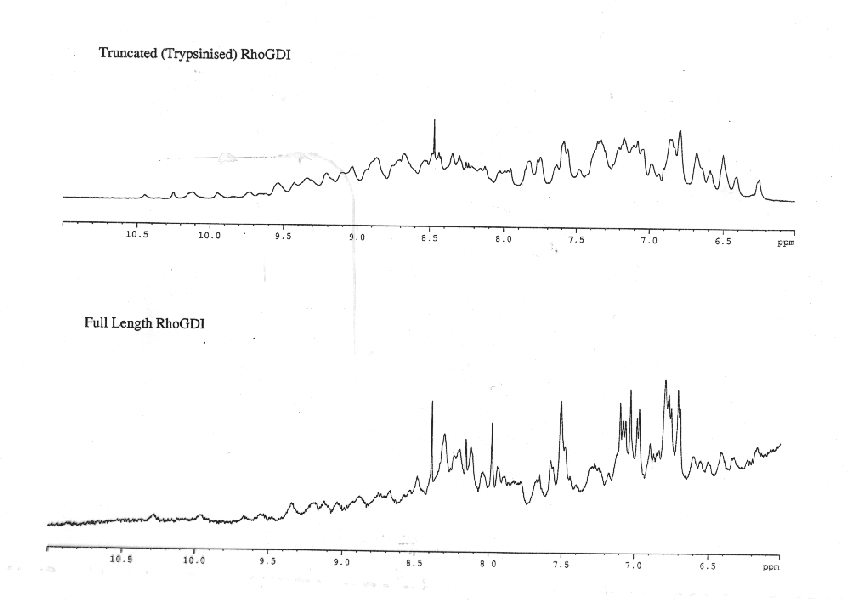
Figure 11. 1D spectra clearly showing the loss of the sharper peaks on proteolytic removal of the first 59 amino acids
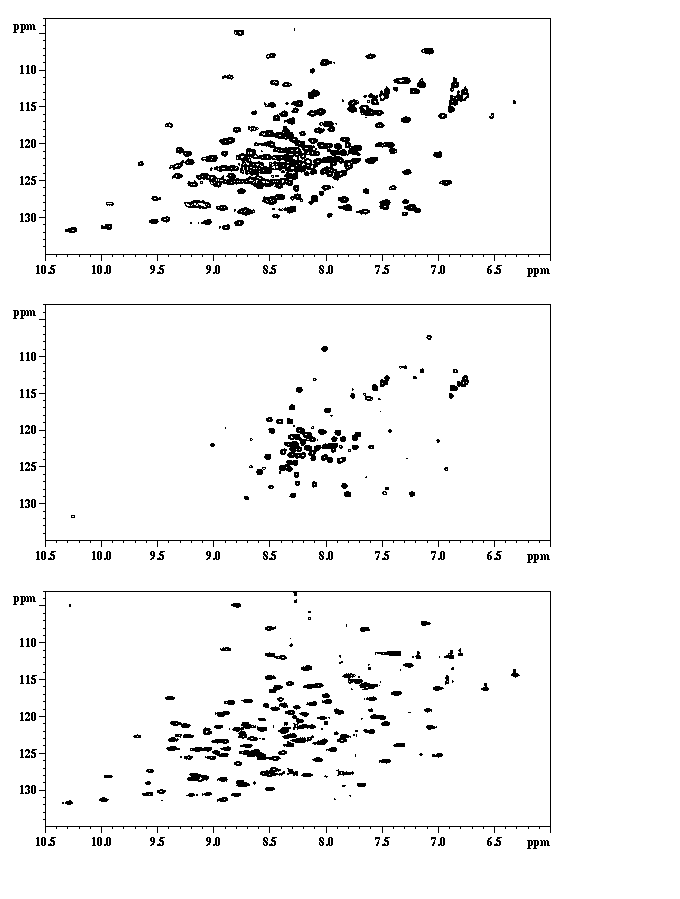
Figure 12. This can also be seen in the 2D 15N-1H HSQC. The top spectrum is the uncleaved. The middle spectra is the same but at a lower sensitivity so only the sharp peaks are seen. The bottom spectrum is after cleavage.
![]()
- http://www.biochem.ucl.ac.uk/~rharris/BCSB/NMR/ NMR within the Bloomsbury Centre for Structural Biology
- UCL/Ludwig Institute NMR pages The NMR groups within the Joint Research School
- http://www.bmrb.wisc.edu/ The Biomagnetic Resonance Data Bank where NMR resonances of proteins are deposited.
- http://neon.chem.le.ac.uk/olderado/olderadohome.html OLDERADO a web site that analyses NMR structure clusters at the University of Leicester.
- 2nd year Chemistry NMR Lectures Henry Rzepa's Lectures from Imperial College
- http://bouman.chem.georgetown.edu/nmr/syllabus.htm Lectures on NMR from Georgetown University USA
- http://www.bruker.com BRUKER a major manufacturers of spectrometers
- http://www.varianinc.com/cgi-bin/nav?varinc/docs/products/nmr/ VARIAN, another major NMR manufacturer
- http://www.jeol.com/nmr/nmr.html JEOL (manufacturer) NMR Website
- NMR for musicians Spectra turned into music?!