Acta Cryst., Vol. D54, (1998), 547-557.

Rfree and the Rfree ratio. Part
I: derivation of expected values of cross-validation residuals used in
macromolecular least-squares refinement
Ian J Tickle,
Roman A Laskowski
&
David S Moss
Department of Crystallography,
Birkbeck College,
Malet Street,
London
WC1E 7HX, UK.
Abstract
The last five years have seen a large increase in the use of cross
validation in the refinement of macromolecular structures using X-ray
data. In this technique a test set of reflections is set aside from the
working set and the progress of the refinement is monitored by the
calculation of a free R-factor which is based only on the excluded
reflections. This paper gives estimates for the ratio of the free
R-factor to the R-factor calculated from the working set for
both unrestrained and restrained refinement. It is assumed that both the
X-ray and restraint observations have been correctly weighted and that
there is no correlation of errors between the test and working sets. It is
also shown that the least-squares weights that minimise the variances of
the refined parameters, also approximately minimise the free
R-factor. The estimated free R-factor ratios are compared
with those reported for structures in the Protein Data Bank.
Introduction
One of the problems in macromolecular crystallography is that the
crystallographer cannot always be sure that an apparently fully refined
structure is free from large systematic errors. The agreement between
the model of the molecular structure and the X-ray diffraction data
from which it has been derived is measured by the crystallographic R
factor, but it is well known that structures with acceptable values of
this parameter can have significant errors (Brändén & Jones,
1990; Kleywegt & Jones, 1995a). The R-factor is susceptible to
manipulation by leaving out weak data or by overfitting the data with
too many parameters and so is not a completely reliable guide to
accuracy. In small-molecule crystallography, where the number of X-ray
intensity observations usually exceeds the number of parameters in the
model by at least an order of magnitude, the R-factor is a more sure
guide to both accuracy and precision.
In 1992 Brünger introduced the idea of an  (Brünger,
1992, 1993), based on the standard statistical modelling technique of
jack-knifing or cross-validatory residuals (McCullagh & Nelder,
1983). The is the same as the conventional R-factor, but
based on a test set consisting of a small percentage (usually ~5-10%) of reflections excluded from a structure refinement. The
remaining reflections included in the refinement are known as the
working set. The value, unlike the R-factor, cannot be
driven down by refining a false model because the reflections on which
it is based are excluded from this process. is only
expected to decrease during the course of a successful refinement.
Consequently, a high value of this statistic and a concomitant low
value of R may indicate an inaccurate model. The procedure assumes
that the reflections removed for the cross-validation test have been
randomly selected and have errors uncorrelated with those that remain
in the set used in the refinement. This assumption may be partly
invalidated by the presence of non-crystallographic symmetry. Ideally,
the refinement should be repeated several times removing
non-overlapping sets of reflections each time.
(Brünger,
1992, 1993), based on the standard statistical modelling technique of
jack-knifing or cross-validatory residuals (McCullagh & Nelder,
1983). The is the same as the conventional R-factor, but
based on a test set consisting of a small percentage (usually ~5-10%) of reflections excluded from a structure refinement. The
remaining reflections included in the refinement are known as the
working set. The value, unlike the R-factor, cannot be
driven down by refining a false model because the reflections on which
it is based are excluded from this process. is only
expected to decrease during the course of a successful refinement.
Consequently, a high value of this statistic and a concomitant low
value of R may indicate an inaccurate model. The procedure assumes
that the reflections removed for the cross-validation test have been
randomly selected and have errors uncorrelated with those that remain
in the set used in the refinement. This assumption may be partly
invalidated by the presence of non-crystallographic symmetry. Ideally,
the refinement should be repeated several times removing
non-overlapping sets of reflections each time.
The is highly correlated with the phase accuracy of the atomic
model (Brünger, 1992, 1993) and can detect various types of errors
in the structure including phase errors and partial mistracing of the
structure. It has also be used in evaluating different refinement
protocols, such as the optimization of the weights used during
refinement. It is particularly useful in preventing the overfitting of
data (Kleywegt & Brünger, 1996).
Kleywegt & Jones (1995a, b) have shown that with low resolution data
it is possible to completely mistrace a structure, deliberately tracing it
backwards through the density, and still achieve an acceptable R
factor. The , on the other hand, could not be duped so easily,
and remained at a high value, close to that expected for a random set of
scatterers, throughout the refinement.
The use of is thus a valuable guide to the progress of
refinement, particularly for low-resolution data, and its use and
publication are widely encouraged. A recent review (Kleywegt & Brünger,
1996) indicated that the use of the measure is becoming more widespread
with it being reported in 44% of articles describing macromolecular X-ray
structures.
However, the usefulness of is limited by the fact that what is
an ``acceptable'' value is often not evident. One would expect
to always be higher than R even when there are no
systematic errors in the model structure, but it is not clear how much
higher it should be. At present we merely have a number of rules of
thumb (Kleywegt & Brünger, 1996).
Cruickshank has estimated that the expected value of the free R-factor
(EFRF) is given by

where  is the number of observations,
is the number of observations,  is the number of
parameters, and R is the conventional R-factor (Dodson, Kleywegt &
Wilson, 1996). Bacchi, Lamzin & Wilson (1996) use this expression in an
extension of the self-validation Hamilton test to assess the significance
of any observed drop in during refinement.
is the number of
parameters, and R is the conventional R-factor (Dodson, Kleywegt &
Wilson, 1996). Bacchi, Lamzin & Wilson (1996) use this expression in an
extension of the self-validation Hamilton test to assess the significance
of any observed drop in during refinement.
The need for more understanding of the behaviour of was
highlighted by Dodson, Kleywegt & Wilson (1996). In spite of the
enthusiasm for its use, actual applications of have
remained somewhat subjective without an understanding of its
statistical basis. For example, if non-crystallographic symmetry (NCS)
constraints are relaxed during a structure refinement, how much should
rise during subsequent refinement if the restrained model
is correct? Without understanding how varies as a function
of the number of restraints and/or number of parameters it is only
possible to make rather subjective judgements.
This paper begins to answer these questions by deriving the expected
value of the free residual from which estimates of both and the
ratio of to R are
calculated.
Theory
Error assumptions
The expected or estimated values of residuals and R-factors will be
derived on the assumption that the weights used in the structure
refinement correctly reflect the errors which include not only
experimental errors in measuring X-ray intensities but also errors in
the functional form of the structure factor model which produce random
and uncorrelated perturbations in the residuals. These model errors,
which may arise from complicated atomic disorder, are an important
source of random error in protein structures and are the reason why
R-factors of refined macromolecular structures are usually higher
than their small molecule counterparts. It is assumed in the
derivation of the statistics in this paper that these random errors
have been correctly accounted for in the weighting of the X-ray data
and of any restraints in the refinement process.
Some model errors, such as the absence of a bulk solvent correction,
lead to correlated model errors in reciprocal space. In the theory to
be presented in this paper, such correlation could be accommodated if
refinement took place with a weight matrix with off-diagonal terms but
in practice computational difficulties preclude the use of such
matrices in macromolecular refinement. All expressions derived in this
paper that use a diagonal weight matrix assume that correlated errors
are absent. Model errors such as missing or misplaced atoms are
similarly assumed to be absent. On the other hand, no assumptions are
made about the completeness or otherwise of the reflection dataset.
In order that the model errors in the free reflections are uncorrelated with
those reflections used in refinement, the reflections in one set must
not be related to those in the other set by crystallographic and
non-crystallographic symmetry. No reflection in the free set must be
related to one in the main set by pseudosymmetry. Care is needed
when selecting reflections from datasets where Bijvoet pairs have been
kept separate. Another case arises when there are domains or molecules
in the asymmetric unit related by a non-crystallographic axis which is
along a rational direction in the crystal lattice (e.g. the
pseudo-dyads in rhombohedral insulin).
Definitions
For convenience the definitions of symbols commonly used in this paper
and its Appendices are grouped together below. A superscript T
denotes a matrix transpose.
Scalars
- f
- the number of structure amplitude observations
included in the refinement (the working set)
- m
- the number of parameters being refined
- n
- the number of observations, including any
restraints, in the refinement
- p
- the number of observations excluded from refinement
(the test set)
- r
- the number of restraints included in the refinement
(r=n-f)
-

- the weight of the ith observation
-

- the standard deviation of the ith observation
-

- an X-ray residual
- D
- the weighted refinement residual
-

- the contribution to D from the working set of f reflections
-

- D based on a test set of p excluded reflections
-

- the contribution to D from the r
restraints (
 )
)
- |F|
- the structure amplitude
- G
- the least-squares X-ray scale factor
-

- the number of atoms being refined
-

- the standard R-factor
-

- the generalised R-factor
-
 &
& 
- R-factors based on all
reflections in the working set
- &

- R-factors based on a test
set of p excluded reflections
Column matrices
-

- the ith row of

-

- the ith row of

- f
- the n observations employed in the refinement (structure
amplitudes and restraints)
- f
- the least-squares estimate of f calculated
at the convergence of the refinement
- g
- the p excluded observations
- g
- the least-squares estimate of g calculated
from
 at the convergence of the refinement
at the convergence of the refinement
- x
- the least-squares estimate of the m parameters
-

- the least-squares residual associated
with the excluded observations
Rectangular matrices
- A
- the least-squares design matrix of derivatives of order n x m
- B
- the p x m matrix analogous to A but involving the excluded observations
-

- the p x p variance-covariance
matrix of the excluded residuals (
 )
)
- H
- the m x m normal matrix given by

- S
- the p x n matrix given by

- W
- the n x n symmetric weight matrix and
 is the VCM (variance-covariance matrix)
of the included observations. This matrix reflects the random experimental and model errors.
is the VCM (variance-covariance matrix)
of the included observations. This matrix reflects the random experimental and model errors.
-

- the p x p symmetric weight matrix of
 and
and  is the VCM of the excluded observations
is the VCM of the excluded observations
The expected value of the free residual
The algebra for the derivation of the expected value of the free
residual is presented in Appendix I. There it is first shown that the
second moment matrix of residuals corresponding to a test set of
observations excluded from least-squares refinement is equal to the
sum of the variance-covariance matrix (VCM) of the omitted
observations and the VCM of the corresponding quantities
calculated from the parameter estimates at the convergence of a
refinement. The expected value of the sum of squared residuals
associated with the excluded observations is then obtained by taking
matrix traces.
The theory then draws on results from an earlier paper (Tickle,
Laskowski & Moss, 1997) where it is shown that when the weighting is
on an absolute scale, the expected value of the sum of a subset of
s weighted residuals at the convergence of a least-squares
refinement is

where the angle brackets denote statistical expectation.
When the above summation is over all n observations (reflections and
restraints)

and hence

In Appendix I equation (21) shows that the expected value of
the residual associated with p excluded observations in the test
set, is given by

It should be noted that the derivation of this equation does
not assume that the test set has been randomly selected from reciprocal
space.
We now consider the f structure amplitude observations
included in the refinement (the working set). From equation
(1) the expected value of the residual
associated with
these observations at convergence is given by

The similarities between equations (3) and
(4) will be noted. In the linear approximation, both
expressions for the expected value are independent of the
observations (structure amplitudes and restraints).
Using these results it is possible to obtain estimates of the ratio of
to R for models with only random uncorrelated
errors. This ratio is estimated first for unrestrained
refinement and then for refinements with geometrical restraints.
These ratios are the starting point for understanding
ratios where systematic errors are present.
Random exclusion of observations in unrestrained refinement
The calculation of the expected values of the residuals using equations
(3) and (4) is a computationally intensive
task in macromolecular refinement, involving the inversion of the
normal matrix H. In general we need statistics which are more
readily available during the refinement process.
We first consider the case of unrestrained refinement. In this case
f=n and from equations (2) and (4)

and

Equation (3) gives the expected value of the free
residual based on any given subset of p observations. In order
to derive simpler expressions, we must now assume that the test
reflections have been randomly chosen from reciprocal space. In this
case when p is large we assume that the sums of the quadratic forms
are approximately proportional to the number of observations
involved. Thus

Hence from equations (3), (5) and (7)

From equations (6) and (8) the
estimated ratio of the residuals for the case of random uncorrelated
errors is

Unlike equations (3) and (4), the
above equation is independent of the scale of the weights.
The expected value of RGfree in unrestrained
refinement
The expected values of the residuals derived in the previous section
may be used to give estimated values of the generalised R-factor,
 , defined by
, defined by

We define  as
as  based on p excluded
reflections. If there are only random and uncorrelated errors then the
numerator of may be approximated by
based on p excluded
reflections. If there are only random and uncorrelated errors then the
numerator of may be approximated by  and using equation (8) we can write
and using equation (8) we can write

We define  as based on all f included
reflections and if the weights have been scaled so that the residual in the
numerator is equal to its expected value then
as based on all f included
reflections and if the weights have been scaled so that the residual in the
numerator is equal to its expected value then

The ratio of RGfree to RG in unrestrained refinement
From equations (9), (10) and (11) we have

Thus we may write the ratio of the generalised R-factors for the
case of random uncorrelated errors as

These results give the expected ratio of the generalised R-factor of
the test set to that of the working set at the convergence of an
unrestrained refinement in the case where there are only random uncorrelated
errors which are correctly reflected in the weights employed. The
results depend only on the number of reflections and the number of
parameters.
RGfree and the RGfree ratio in
restrained refinement
In a restrained refinement, such as is typical in macromolecular
crystallography, the ratios derived in the previous two sections would
only be applicable if were calculated from a random
selection of residuals including both structure amplitude observations
and restraints. Since R-factors are traditionally only based on structure
amplitudes, the estimation of ratios for restrained
refinement requires further analysis.
The number of observations this time is n and f of these are
structure amplitudes, the balance consisting of r geometrical,
thermal or other restraints which make a contribution to
the minimised residual at convergence. From equation (1)
we have

where the summation is taken over the restraint observations. Using
this equation and equation (2) the summation over the
structure amplitude observations can be written

This result and equations (4) and (7) give the following
approximation for

Similarly an approximation to  can be derived
from equations (4), (7) and (14).
can be derived
from equations (4), (7) and (14).

Hence an estimate of at the convergence of a
correctly weighted restrained refinement with only random uncorrelated
errors is

The estimated ratio of the free residual to the included
residual is given by

and hence

Minimum variance weights minimise Rfree
The expected values derived above are only applicable for correctly weighted
least-squares refinements. However has also been used to
optimise the weighting of geometrical or temperature factor terms in
refinement by adjusting the weights so as to minimise
(Brünger, 1992, 1993). It is therefore of interest to enquire how
responds to variations in weighting.
Appendix III shows that the weights  which correctly reflect
experimental and model errors, minimise the variance of both the
refined parameters , and also the expected value of the
sum of the squares of the unweighted residuals in the test set. Hence
the choice of these weights approximately minimises . One
method of estimating such weights has been described by Tickle,
Laskowski & Moss (1998).
which correctly reflect
experimental and model errors, minimise the variance of both the
refined parameters , and also the expected value of the
sum of the squares of the unweighted residuals in the test set. Hence
the choice of these weights approximately minimises . One
method of estimating such weights has been described by Tickle,
Laskowski & Moss (1998).
The use of standard R-factors
The R-factor expressions used so far in this paper have been based
on the generalised R-factor, . However identical expressions
can be derived for the estimated values of the standard free R-factor,
, and the standard ratio. For example, in
Appendix II the following estimate of the ratio in
unrestrained refinement is derived which is based on standard
R-factors.

The numerator and denominator in the above equation are
first order binomial approximations to square roots and therefore
another estimate of the standard ratio is

The right hand side is now the same as for the ratio
(equation 12). These expressions may be compared with
the estimate used by Bacchi, Lamzin & Wilson (1996) which in our
notation is

The derivation of simple estimates for the standard and its
ratio requires the assumption that the variances of all structure
amplitudes are equal. The use of generalised R-factors, whose
estimates do not require this assumption, is to be preferred.
Discussion
Restrained and unrestrained RGfree ratios
has been employed (Kleywegt & Brünger, 1996) to detect
situations where addition of extra parameters to a refinement brings
no significant improvement in the model. For example the extra
parameters may concern the relaxation of non-crystallographic symmetry
or the employment of individual atomic temperature factors. Relevant
estimates of are summarised in
Table 1 and
help to make these discussions more quantitative. The Table gives the
more important expressions derived earlier for a refinement at
convergence with only random uncorrelated errors and weighting which
correctly reflects the model and experimental errors.
The constrained and unrestrained statistics in the Table show what
happens when constraints are imposed, assuming that the reduction in
parameters does not introduce model error. It is seen that a reduction
in the expected values of and the ratio takes
place. A reduction also takes place when restraints are imposed,
assuming again that the above conditions are not violated.
The meaning of the Rfree ratio
The estimated ratios derived in this paper are the values
that should be achievable at the end of a structure refinement when
only random uncorrelated errors exist in data and model provided that
the observations have been properly weighted (see below).
A larger ratio than that predicted by these formulae may
indicate that parameter shifts have taken place which have minimised
the residual without significantly improving the model. This may arise
when errors in the model are sufficiently large for the refinement to
descend into a false minimum.
A smaller ratio than that predicted by these formulae may
indicate that the refinement has not reached convergence since the
initial value of the ratio immediately after the division of the data
into a working set and a test set will be approximately
unity. Interpretation requires care since a wrong model which has not
been fully minimised against the data may produce the same ratio as a
fully minimised correct model.
In macromolecular refinement, model error is usually the major
contributor to R and at the end of the
refinement. Paucity of diffraction data means that thermal and static
disorder in the more mobile parts of the molecule cannot be accurately
modelled. These model errors may cause random perturbations in the
structure amplitude residuals which are indistinguishable from random
experimental errors. The ratio will not be affected by the
magnitude of these errors provided that the latter have independent
effects on the included and excluded residuals.
Observed Rfree ratios from the Protein Data Bank
We have examined ratios for crystal
structures in the Protein Data Bank (Bernstein et al, 1977) as at 1 June
1997. Figure 1 shows a plot of the
ratio as a function of  , where is the number of atoms included in the refinement and
f is the number of reflections used, for 725 macromolecular
structures for which all these values are reported. The points are colour
coded according to resolution range.
, where is the number of atoms included in the refinement and
f is the number of reflections used, for 725 macromolecular
structures for which all these values are reported. The points are colour
coded according to resolution range.
We define the ratio as  and
and
 . Values of y range from about 0.8 to 1.8. By
substituting
. Values of y range from about 0.8 to 1.8. By
substituting  into equation (15), we have
into equation (15), we have

Figure 1 shows the curves
corresponding to equation (16) for different
values of a.
In order to make the comparison between experimental and theoretical values
easier, a function of y was sought which is a linear function of
x. By squaring and rearranging the terms in equation (16) we arrive at

where

Figure 2 shows a plot of
z against x where the points are colour coded as in Figure 1. The coloured straight lines
in Figure 2 are least-squares
lines fitted to the data points in the particular resolution range
represented by points of the same colour. For example, the pink triangular
points represent data between 3 and 4Å\ resolution and the pink line
is the least-squares line through the pink triangular points. The pecked
black lines emanating from the origin in Figure 2 are plots of
z=ax for the same values of a as shown in Figure 1.
We requested information from some of the authors whose structures were
outliers in Figure 2. It became
apparent that very unusual ratios are normally not the
result of careful refinement protocols. The coloured lines were therefore
plotted ignoring the points in the darker regions outside the sector
bounded by the black lines of slopes 0.5 and 10. The choice of these slopes
as cutoffs was somewhat arbitrary but the removal of these outliers caused
the coloured lines to pass nearer to the origin.
The plots of z=ax represent refinement regimes with different
numbers of parameters per atom. The gradients of the lines (a)
increase with the number of parameters per atom. In the absence of
relevant information in the Data Bank, it was assumed that in
restrained refinements,  and
and  . These estimates
ignore temperature factor restraints (if any) because our survey of
the latter revealed widely different restraint protocols. Using these
values, for restrained refinements
. These estimates
ignore temperature factor restraints (if any) because our survey of
the latter revealed widely different restraint protocols. Using these
values, for restrained refinements

It can be seen that the z=2x line (isotropic temperature factors)
passes through the constellations of orange crosses and pink
triangular points, representing structures between 2.5 and 1.5Å\
resolution and is close to the green pecked line (2.5-2.0Å\
data). Similarly the z=x line (overall temperature factor) lies
close to the pink line which is fitted to the 4 to 3Å data. Even in
the absence of details of restraint procedures, the z=ax lines can
be seen to pass through areas of the plot where the particular
refinement regime is most relevant. The large spread of values about
the straight lines is unlikely to be solely a statistical effect and
may well say something about the quality of the refinements.
Comparison of the lines z=2x and z=4x, which differ only in
respect of restraints, shows how restraints lower the
ratio. Non-crystallographic symmetry (NCS, see Introduction) might
give rise to lower than predicted ratios. However, a check
on structures in our plots which exhibit NCS, did not reveal any
obvious systematic effects.
Conclusion
Values of are affected by all types of error in the model
and the data. The ratio, however, is independent of random
errors and provides a statistic which can be compared with its
theoretically estimated value and used to detect systematic model or
weighting errors at the convergence of least-squares refinement.
However, achievement of a theoretical value of the ratio is not by
itself proof of the correctness of the model or of the quality of the
refinement. Nevertheless it would still be helpful if refinement
programs printed out the calculated and estimated value of the
ratio using the expressions shown in Table 1. This would
encourage a better understanding of than exists at
present. Calculation of the observed and theoretical values of these
ratios has already been implemented in the refinement program
RESTRAIN (Driessen et al., 1989).
At low resolution the number of data excluded for cross-validation may
be small and in these circumstances the precision of free residuals is
important. This will be the subject of part II of this work.
The authors would like to thank Professor D W J Cruickshank FRS for his
helpful comments on this paper.
Statistical properties of free residuals
Derivation of the expected value of the free residual
Here we derive the statistical expectation of the free residual , at the convergence of a least-squares refinement.
The normal equations of least-squares refinement at convergence can be
written
If the errors in the observations and the model are not too large
then a truncated Taylor expansion may be written about the expected values of the parameter vector
 and the observation vector
and the observation vector  .
.

The structure amplitudes and target distances corresponding
to the excluded observations in a test set can be expressed in terms
of the parameter estimates by the truncated Taylor expansion

Thus the column of residuals of the excluded observations is given by

Assuming that the errors in  and are
uncorrelated, the VCM of the residuals associated with the excluded observations is given by
and are
uncorrelated, the VCM of the residuals associated with the excluded observations is given by

Thus is equal to the sum of the VCM of the
excluded observations and the VCM of the corresponding
quantities calculated from the refined parameters. From equation
(18) it follows that

where  is a unit matrix of order p. Using the fact
that
is a unit matrix of order p. Using the fact
that

and noting that the trace of a product of matrices is
invariant under a cyclic permutation of the order of matrix
multiplication, we take the trace of both sides of equation
(19) to give

If the p excluded observations are structure amplitudes
and we assume that the weight matrix is diagonal, then equation
(20) can be written as

where the angle brackets denote statistical expectation, is the expected value of the residual associated with
the given p excluded observations and is the i th
row of B. The expected value of a single weighted excluded
residual in the summation is obtained by taking a single diagonal term
from equation (19) which gives

Two notable assumptions have been made in the above analysis. First,
it has been assumed that the test set residuals are uncorrelated with
those in the working set. Equation (18) is invalid if this
assumption is not true.
Second, it is assumed that the refinement has used a weight matrix
which correctly reflects the experimental and model
errors. Equation (21) is invalid if this assumption is not
true. When a diagonal weight matrix is used, as is almost always the
case in practice, correlated errors in reciprocal space will not be
correctly represented. In Appendix III it is
shown that the use of
weight matrices which do not correctly account for the errors will
lead to test set residuals with larger variance-covariance matrices.
The ratio of Rfree to R in unrestrained
refinement
The derivations in the body of this paper have been given in
terms of the generalised R-factor. In this Appendix we derive the
expected values of residuals expressed as the sum of unweighted absolute
differences. Hence we obtain an estimate of the ratio
expressed in terms of the standard R-factor which is defined as

The variance of an included residual at the convergence of a refinement (see Appendix II
in Tickle, Laskowski and Moss, 1997) can be written as

To make further progress we need to assume a distribution for each
residual  . If a normal distribution
is assumed, we can write
. If a normal distribution
is assumed, we can write

and hence

The square root in the above summation may be expanded binomially to the first order giving

We now assume that is the same for all observations so that we can write

Using equation (5) to simplify the right-hand side, we can write

By analogous reasoning the sum over the p excluded reflections can be approximated as

We have now derived approximations for the numerators of and
. Their denominators are, on average, proportional to the
number of reflections in the respective summations. Hence dividing
equation (24) by equation (23) and
multiplying by f/p we have
Minimum variance weights minimise the expected value of
Rfree
First we show that the least-squares refinement using
a weight matrix which is the inverse of the VCM of the
observations, minimises the VCMs of both the refined parameters and the free residuals  .
.
Consider a function  which is a function of the
refined least-squares parameters which is linear to within a first
order Taylor approximation.
which is a function of the
refined least-squares parameters which is linear to within a first
order Taylor approximation.

Now consider two column matrices  and
and  , defined
below, which are both unbiased estimates of .
, defined
below, which are both unbiased estimates of .

where

 is any weight matrix and
is any weight matrix and  . From
equations (27) and (28) and from
the definitions of
. From
equations (27) and (28) and from
the definitions of  and
and 

We wish to show that the VCM of is smaller than that of . Because and are unbiased estimators,  and thus the VCM of can be
expressed as
and thus the VCM of can be
expressed as

The last two terms of the above equation are the transpose of each other
and are each zero matrices as shown by the following analysis which uses
equations (25), (26), (27) and (29).

Hence from equation (30)

Since the VCMs are positive definite

Thus the VCM of which is calculated with is less than the VCM of which is calculated with
another weight matrix . Making the substitution  and setting
and setting  to a unit matrix, this analysis shows
that by using the weight matrix , we minimise the variance of
. Substituting
to a unit matrix, this analysis shows
that by using the weight matrix , we minimise the variance of
. Substituting  and
and  , the same analysis shows that also minimises the VCM of
, the same analysis shows that also minimises the VCM of
 . From equation (18) the VCM of the residuals
associated with the excluded observations is the sum of the
constant matrix and the VCM of . Hence
and its trace are also minimised by choosing as the
weight matrix.
. From equation (18) the VCM of the residuals
associated with the excluded observations is the sum of the
constant matrix and the VCM of . Hence
and its trace are also minimised by choosing as the
weight matrix.
The trace of is the expected value of the unweighted sum of
squared residuals

where the summation is taken over the p reflections in the test set. By
using the normal approximation in equation (22) we can say
that the sum of absolute differences

and hence are approximately minimised by choosing as the
least-squares weight matrix.
- Bacchi A, Lamzin V S & Wilson K S (1996). A self-validation
technique for protein-structure refinement: the extended Hamilton test.
Acta Cryst., D52, 641-646.
- Bernstein F C, Koetzle T F, Williams G J B, Meyer E F Jr,
Brice M D, Rodgers J R, Kennard O, Shimanouchi T & Tasumi M. (1977). The
Protein Data Bank: a computer-based archival file for macromolecular
structures. J Mol Biol 112: 535-542.
- Brändén C-I & Jones T A (1990). Between objectivity and
subjectivity. Nature, 343, 687-689.
- Brünger A T (1992). Free R value: a novel statistical
quantity for assessing the accuracy of crystal structures. Nature,
355, 472-475.
- Brünger A T (1993). Assessment of phase accuracy by cross
validation: the free R value. Methods and applications. Acta
Cryst., D49, 24-36.
- Dodson E, Kleywegt G J & Wilson K (1996). Report of a workshop on
the use of statistical validators in protein X-ray crystallography.
Acta Cryst., D52, 228-234.
- Driessen H, Haneef M I J, Harris G W, Howlin B, Khan G & Moss D S
(1989). RESTRAIN: restrained structure-factor least-squares refinement
program for macromolecular structures. J. Appl. Cryst., 22,
510-516.
- Kleywegt G J & Brünger A T (1996). Checking your imagination:
applications of the free R value. Structure, 4, 897-904.
- Kleywegt G J & Jones T A (1995a). Where freedom is given, liberties
are taken. Structure, 3, 535-540.
- Kleywegt G J & Jones T A (1995b). Braille for pugilists. In
Making the Most of Your Model, eds. Hunter W N, Thornton J M & Bailey S,
SERC Daresbury Laboratory, Warrington, UK, pp. 11-24.
- McCullagh P & Nelder J A (1983). Generalised Linear
Models. pub. Chapman & Hall, London, UK, pp. 209-221.
- Tickle I J, Laskowski R A & Moss D S (1998). Error estimates of
protein structure coordinates and deviations from standard geometry by full
matrix refinement of
 B- and
B- and  B2-crystallin. Acta
Cryst., D54, 243-252.
B2-crystallin. Acta
Cryst., D54, 243-252.
Figures
Figure 1
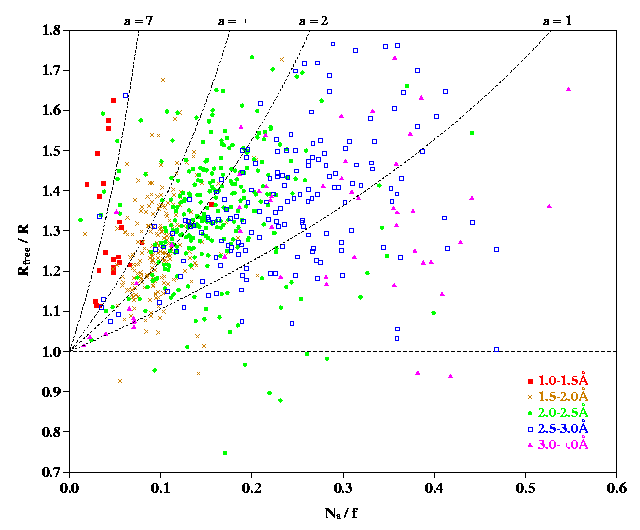 PostScript
PostScript
Figure 1. Plot of the  ratio as a function of the ratio for 725
macromolecular structures in the Protein Data Bank, where
ratio as a function of the ratio for 725
macromolecular structures in the Protein Data Bank, where  is the number of atoms
included in the refinement and f the number of reflections used. The
data points are colour-coded according to their resolution range, as shown
in the key on the bottom right of the graph. The high resolution data
points tend to be close to the vertical axis with the other points tending
to spread further to the right the lower their resolution, as might be
expected. Also shown are four dotted curves corresponding to different
values of the variable a. The curves shown are for: a = 1
which corresponds to 3 parameters per atom (ie restrained
refinement of atomic coordinates only, plus an overall temperature factor);
a = 2 is for 4 parameters per atom (restrained refinement of
coordinates plus individual isotropic temperature factors); a = 4
represents unrestrained refinement with 4 parameters per atom; and a
= 7 represents 9 parameters per atom (restrained anisotropic refinement).
is the number of atoms
included in the refinement and f the number of reflections used. The
data points are colour-coded according to their resolution range, as shown
in the key on the bottom right of the graph. The high resolution data
points tend to be close to the vertical axis with the other points tending
to spread further to the right the lower their resolution, as might be
expected. Also shown are four dotted curves corresponding to different
values of the variable a. The curves shown are for: a = 1
which corresponds to 3 parameters per atom (ie restrained
refinement of atomic coordinates only, plus an overall temperature factor);
a = 2 is for 4 parameters per atom (restrained refinement of
coordinates plus individual isotropic temperature factors); a = 4
represents unrestrained refinement with 4 parameters per atom; and a
= 7 represents 9 parameters per atom (restrained anisotropic refinement).
 PostScript
PostScript
Figure 2. Plot of the same data as in Fig.1 but with the vertical
axis representing z which is a linear function of  , where
, where  and
and  . Here the curves
from Fig.1, which corresponded to different values of a, are now
straight lines. Again, the data points are colour-coded according to
resolution range, as shown in the key on the right. Also shown are 5
coloured lines representing least-squares lines fitted to the data points
in each of these five different resolution ranges. The lines are identified
by the appropriate point-markers in square brackets outside the graph
border.The grey regions show the regions from which the data points have
been excluded in the least-squares lines calculations, namely points
outside the sector bounded by the lines corresponding to a = 10 and
a = 0.5.
. Here the curves
from Fig.1, which corresponded to different values of a, are now
straight lines. Again, the data points are colour-coded according to
resolution range, as shown in the key on the right. Also shown are 5
coloured lines representing least-squares lines fitted to the data points
in each of these five different resolution ranges. The lines are identified
by the appropriate point-markers in square brackets outside the graph
border.The grey regions show the regions from which the data points have
been excluded in the least-squares lines calculations, namely points
outside the sector bounded by the lines corresponding to a = 10 and
a = 0.5.
Table 1

Document created using latex2html